Chondro-osseous tumours
Chondro-osseous tumours occasionally cross the desk of the pathologist. They are grouped together as bone may develop from cartilage.
Primary bone tumours are rare; the most common bone tumour is metastases.[1]
Bone tumours occasionally are lumped with soft tissue tumours. Soft tissue tumours are dealt with in the soft tissue lesions article. An introduction to bone is found in the bone article. An introduction to cartilage is found in the cartilage article.
General
- Diagnosis of a primary bone tumour should not be made without radiologic & clinical information!
- Metastasis:primary bone tumours = >20:1.[1]
Common malignant
- Osteosarcoma.
- Chondrosarcoma.
- Ewing's sarcoma.
- Multiple myeloma.
- Metastases.
- Most common tumours metastatic to bone (mnemonic: BLT with Ketchup & Pickles):
Epidemiology:[2]
- Osteosarcoma -> 2nd decade.
- Ewing's ->5-20 yrs.
- Chondrosarcoma -> from enchondroma or osteochrondroma -- patients over 40 yrs.
- Multiple myeloma -> most common primary bone tumour in adults.
Malignant bone tumours by age
Most common by age:[3]
- <1 year old - neuroblastoma.
- 1-10 years old - Ewing's of tubular bones.
- 10-30 years old - osteosarcoma, Ewing's of flat bones.
- 30-40 years old - reticulum cell sarcoma, fibrosarcoma, parosteal osteosarcoma, malignant giant cell tumour, lymphoma.
- >40 years old - mets, multiple myeloma, chondrosarcoma.
Benign aggressive bone tumours
- Giant cell tumours.
- Osteoblastoma.
- Thought to be related to osteoid osteoma.
- If in long bones often diaphyseal.
Cartilage
Enchondroma
General
- Benign thingy.
- Usu. legs and feet.
- May be difficult to separate from chondrosarcoma.
- Multiple chondromas = enchondromatosis; three distinct syndromes.[6]
Radiology:[6]
- Lytic lesion.
- Usu. close to a growth plate.
Clinical:[6]
- Pain.
Microscopic
Features:
- Ctyologically benign cells is spaced nests.
Images:
{kind=link}
{kind=link}
Chondroblastoma
General
- Growth plate lesion.
- Sclerotic margin.
- "Young" = growth plates open.
Microscopic
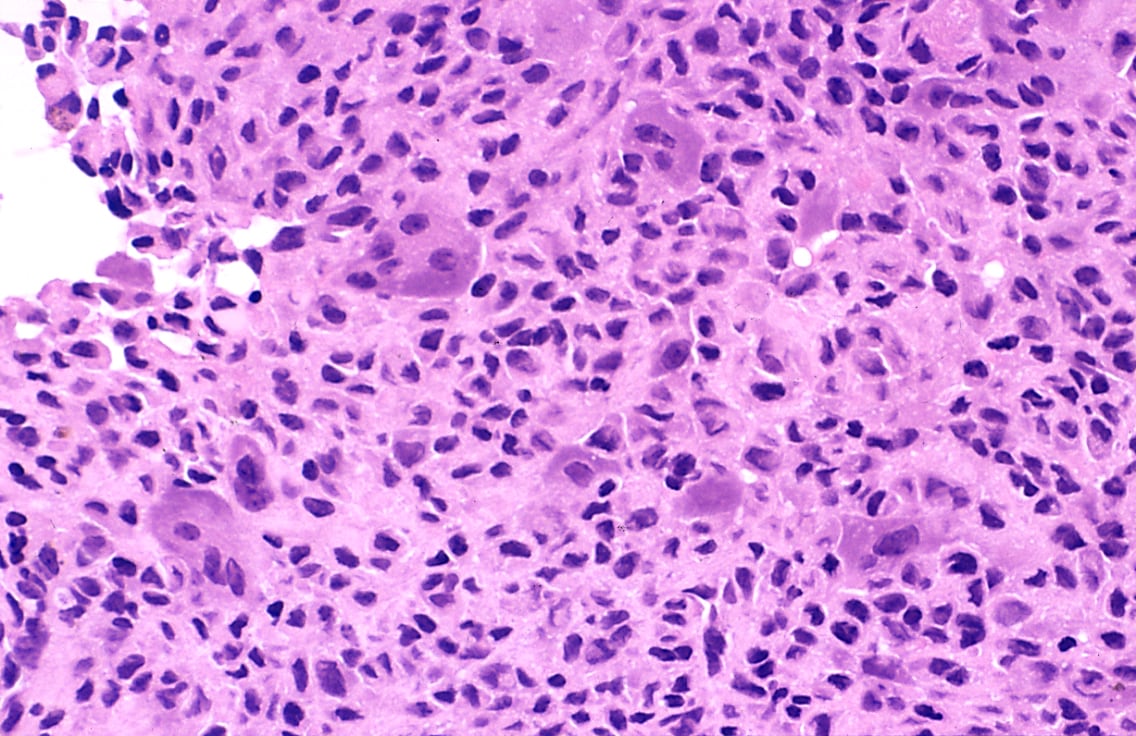
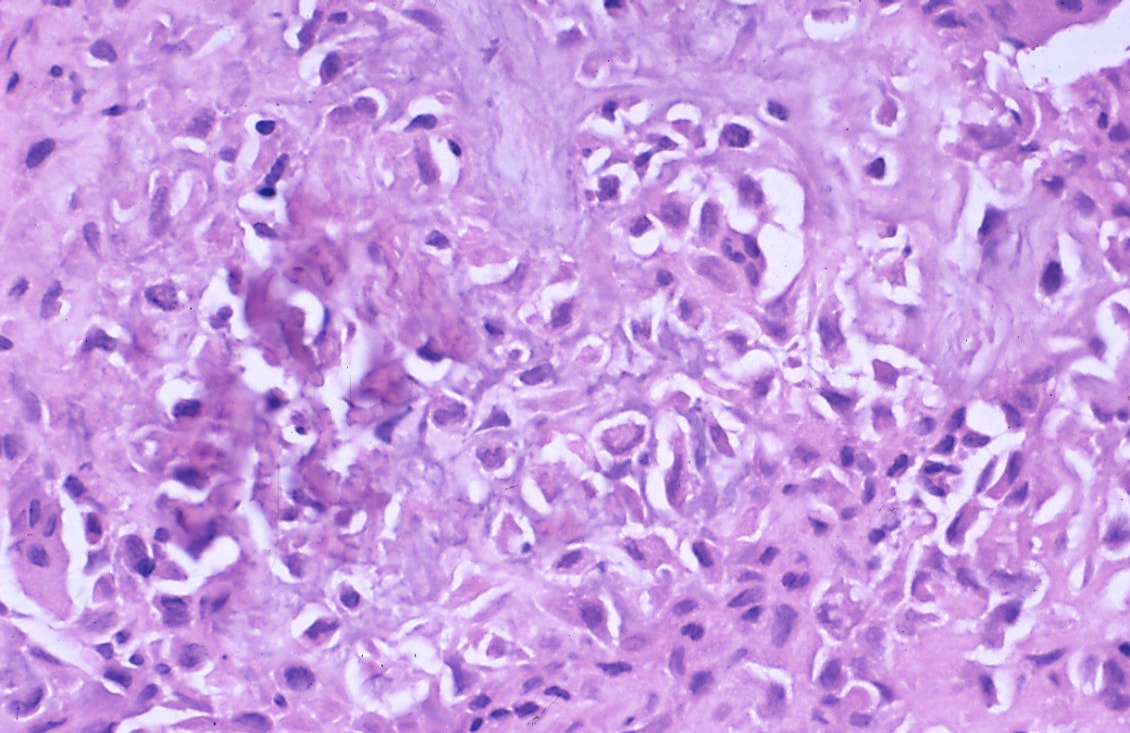
Features:[7]
- Abundant extracellular material - pink on H&E stain - looks vaguely like cartilage.
- Chondroblasts:
- Nuclear morphology variable: ovoid, folded or grooved.
- Moderate-abundant eosinophilic cytoplasm.
- +/-Calcifications surround cells nests ("chickenwire" appearance) - classic feature.
- +/-Giant cells.
- May lead to confusion with giant cell tumour.
Images:
- Chondroblastoma - intermed. mag. (WC).
- Chondroblastoma - very high mag. (WC).
- Chondroblastoma (medscape.com).[8]
- Chondroblastoma with "chickenwire" appearance (medscape.com).[8]
{kind=link}
{kind=link}
{kind=link}
{kind=link}
IHC
Features:[7]
- S100 +ve.
- Vimentin +ve.[8]
Chondrosarcoma
General
- Usually a good prognosis.
Clinical/epidemiologic features:[9]
- Usually arise in a (benign) abnormality of cartilage (e.g. osteochondroma, enchondroma).
- May be associated with a syndrome:
- Olier disease (multiple enchondromatosis).
- Maffucci syndrome (multiple enchondromas and hemangiomas).
Notes:
- Review article (from oncology perspective): PMID 17545802.
Subtypes
Several subtypes exist:
- Chondrosarcoma not otherwise specified (NOS).
- Juxtacortical chondrosarcoma.
- Myxoid chondrosarcoma.
- Mesenchymal chondrosarcoma.
- Clear cell chondrosarcoma.
- Dedifferentiated chondrosarcoma chondrosarcoma.
Microscopic
- "Abnormal cartilage" (esp. low power):
- Nuclear atypia.
- Nuclear clearing.
- Nucleoli.
- Nuclear atypia.
Notes:
- More cellular than cartilage... but relatively paucicellular compared to other sarcomas.
Images:
.jpg){kind=link}
.jpg){kind=link}
.jpg){kind=link}
DDx:
Variants
Mesenchymal chondrosarcoma
- Arise in soft tissue; this is where the name comes from.[12]
- Rare variant of chondrosarcoma.
Microscopic: Features:
- "White clouds in a blue sky".
Image:
Grading
Features:[13]
- Grade I: moderate cellularity +/- binucleated cells.
- Grade III: nuclear pleomorphism, mitoses common.
- Grade II: between Grade I and Grade III.
IHC
- S-100 -ve. (???)
Bone
Osteoid osteoma
General
- Benign bone lesion.
Clinical:[14]
- Extremely painful.
- Relieved by NSAIDS.
Microscopic
Features:[14]
- Anastomosing bony trabeculae with:
- Variable mineralization.
- Mineralization (calcium phosphate) = purple on H&E stain.
- Osteoblasts rimming.
- Cells line-up at edge of bone.
- Variable mineralization.
Images:
- Osteoid osteoma - CT scan (med.utah.edu).
- Osteoid osteoma (sciencephoto.com).
- Osteoblastoma - high mag. (WC) - histological similar to OO.
- Osteoblastoma - low mag. (WC) - histological similar to OO.
{kind=link}
{kind=link}
Notes:
- Histomorphologically near identical/indistinguishable from osteoblastoma.[15]
Osteoblastoma
General
- Benign bone tumour.
Microscopic
Features:[14]
- Anastomosing bony trabeculae with:
- Osteoblasts rimming.
- Cells line-up at edge of bone.
- Osteoblasts rimming.
Notes:
- Histomorphologically near identical/indistinguishable from osteoid osteoma.[15]
- Must be greater 1.5 cm by definition.[15]
Images:
Ewing sarcoma
- AKA EWS/PNET:
- EWS = Ewing sarcoma.
- PNET = (peripheral) primitive neuroectodermal tumour.
- EWS and PNET were once thought to be different tumours.
Notes:
- Peripheral primitive neuroectodermal tumour should not be confused with primitive neuroectodermal tumour a (supertentorial) brain tumour with similarities to medulloblastoma.
General
Clinical
- Painful.
- Usually younger than 20 years.
- Second most common malignant bone tumour in children.
- Most common malignant bone tumour = osteosarcoma (AKA osteogenic sarcoma).
Poor prognostic factors:[16]
- Age (18 years-old+).
- Pelvis (extremity = good).
- >8 cm.
- Metastases.
- EWS-FL1 fusion type 2.
Radiology
Features:[17]
- Long bones, diaphyses.
- Destructive.
- "Onion-skin" periosteal reaction.
Microscopic
Features:[18]
- Scant clear cytoplasm (contain glycogen -- PAS +ve, PAS-D -ve).
- Bland round small nucleus.
- Usu. lack nucleoli.
- Usu. minimal-moderate size variation.
Notes:
- It is a small round cell tumour.
IHC
Features:[19]
- CD99 +ve -- 1. diffuse, 2. plasma membrane staining; both required -- most specific.
- CD45 -ve.
- Done to r/o lymphoma.
- +/-Neural markers (NSE, synaptophysin, CD57 (??? CD56 ???), S100).
- +/-Cytokeratins.
- Caveolin-1 +ve in ~ 85% of EWS.[20]
Notes:[21]
- CD99 +ve (plasma membrane) tumours:
- Lymphoblastic lymphoma/leukemia.
- Angiomatoid fibrous histiocytoma.
- Desmoplastic small round cell tumour.
Molecular diagnostics
Common features:
- EWS/FLI-1 fusion gene formation due to translocation: t(11;22)(q24;q12).[22][23]
- Often detected by RT-PCR (with EWS 5' and FLI-1 3' primers).
- Type 1 = EWS exon 7 + FLI-1 exon 6; good prognosis.
- Type 2 = others; poor prognosis.
Notes:
- The t(11;22)(q24;q12) is seen in ~90% of EWS/PNET... but also in:
- Olfactory neuroblastoma.
- Small cell osteogenic sarcoma.
- Polyphenotypic tumours.
- Rhbdomyosarcoma.
- Neuroblastoma (possibly).
- Several other translocations exist.
- Lack of molecular findings does not exclude Ewing sarcoma.
Osteosarcoma
- AKA osteogenic sarcoma.
General
- Most common malignant bone tumour in children.
Trivia:
- Terry Fox was afflicited by this tumour.
Definition
- Tumour that makes osteoid.
- Osteoid = (extracellular) organic component of bone, normally produced by osteoblasts (cells which make bone matrix).
Microscopic
Features:
- Cells with malignant features (e.g. nuclear membrane irregularities, marked nuclear size differences, mitoses) surrounded by delicate strands of osteoid.
- Osteoid on H&E: pink, homogenous, "glassy".
- Tumours typically very cellular - when compared to normal bone.
- Large (multinucleated) osteoclast-like giant cells may be seen.[24]
Images:
{kind=link}
{kind=link}
Subtypes
- Many subtypes exist.
- Conventional osteosarcoma (high grade).
- Osteoblastic.
- Fibroblastic - MFH-like.
- Chondroblastic - may be confused with chondrosarcoma.
- Small cell.
- Telangiectatic - extremely vascular.
- Parosteal.
- Periosteal.
- Low grade central.
- High grade surface.
- Secondary.
- Gnathic - jaw bones; usu. chondroblastic.
Other
Diffuse tenosynovial giant-cell tumour
- AKA tenosynovial giant-cell tumour, diffuse type.
- Previously known as pigmented villonodular synovitis (PVNS).[27]
General
- Course: benign.
- Giant cell tumor of the tendon sheath is considered to be the soft-tissue counterpart of PVNS.[28]
Microscopic
Features:[29]
- Subsynovial nodules composed of cells with:
- Abundant cytoplasm.
- Pale nuclei.
- Multinucleated giant cells.
- Hemosiderin-laden macrophages.
- Foam cells.
Images:
{kind=link}
{kind=link}
Adamantinoma
General
Features:[17]
- Rare: < 1% of bone tumours.
- 25-35 years old.
- Tibia, fibula.
- Benign, may be locally aggressive.
- Cousin of ameloblastoma. (???)
Radiology
- Intracortical, radiolucent.
Microscopic
Features:
- Biphasic tumour:
- Fibrous/spindle cell component.
- Epithelial component.
Images:
{kind=link}
Brown cell tumour
Etiology
- Due to hyperparathyroidism - usually parathyroid adenoma.
- Usually secondary to chronic renal failure.
- Not a true neoplasm,[31] i.e. the name is a misnomer.
- May (clinically) mimic a true neoplasm.
Microscopy
Features:
- Fibrosis.
Hypercalcemia DDx
Mnemonic GRIMED:[32]
- Granulomatous disease (tuberculosis, sarcoidosis).
- Renal disease.
- Immobility.
- Malignancy (esp. squamous cell carcinoma, plasmacytoma).
- Endocrine (primary hyperparathyroidism - leads to brown cell tumour).
- Drugs (thiazides ... others).
Giant cell tumour of bone
General
Features:[33]
- Approximately 5% of primary bone tumours.
- Typical age: 20-45 years.
Clinical
- May present with joint pain, immobility.
Note:
- Several types of giant cell tumours exist.
Microscopic
Features:[34]
- Giant cells with abundant nuclei (usu. >10 in the plane of section).
- Usu. have prominent nucleoli.
- Mononuclear cells and small multinucleated cells with nuclei similar to those in the giant cells - key feature
Images:
{kind=link}
{kind=link}
See also
References
- ↑ 1.0 1.1 Humphrey, Peter A; Dehner, Louis P; Pfeifer, John D (2008). The Washington Manual of Surgical Pathology (1st ed.). Lippincott Williams & Wilkins. pp. 632. ISBN 978-0781765275.
- ↑ TN05 OR42.
- ↑ TN05 OR42.
- ↑ TN05 OR41.
- ↑ URL: http://www.emedicine.com/RADIO/topic494.htm.
- ↑ 6.0 6.1 6.2 URL: http://emedicine.medscape.com/article/389224-overview. Accessed on: 25 December 2010.
- ↑ 7.0 7.1 Humphrey, Peter A; Dehner, Louis P; Pfeifer, John D (2008). The Washington Manual of Surgical Pathology (1st ed.). Lippincott Williams & Wilkins. pp. 642. ISBN 978-0781765275.
- ↑ 8.0 8.1 8.2 URL: http://emedicine.medscape.com/article/1254949-diagnosis. Accessed on: 31 December 2010.
- ↑ Skubitz KM, D'Adamo DR (November 2007). "Sarcoma". Mayo Clin. Proc. 82 (11): 1409–32. PMID 17976362. http://www.mayoclinicproceedings.com/content/82/11/1409.long.
- ↑ IAV. 26 February 2009.
- ↑ Klatt, Edward C. (2006). Robbins and Cotran Atlas of Pathology (1st ed.). Saunders. pp. 417. ISBN 978-1416002741.
- ↑ Dowling EA (June 1964). "Mesenchymal chondrosarcoma". J Bone Joint Surg Am 46: 747–54. PMID 14161087. http://www.ejbjs.org/cgi/reprint/46/4/747.pdf.
- ↑ Humphrey, Peter A; Dehner, Louis P; Pfeifer, John D (2008). The Washington Manual of Surgical Pathology (1st ed.). Lippincott Williams & Wilkins. pp. 643. ISBN 978-0781765275.
- ↑ 14.0 14.1 14.2 Mills, Stacey E; Carter, Darryl; Greenson, Joel K; Oberman, Harold A; Reuter, Victor E (2004). Sternberg's Diagnostic Surgical Pathology (4th ed.). Lippincott Williams & Wilkins. pp. 285. ISBN 978-0781740517.
- ↑ 15.0 15.1 15.2 Mills, Stacey E; Carter, Darryl; Greenson, Joel K; Oberman, Harold A; Reuter, Victor E (2004). Sternberg's Diagnostic Surgical Pathology (4th ed.). Lippincott Williams & Wilkins. pp. 286. ISBN 978-0781740517.
- ↑ PST. 14 February 2011.
- ↑ 17.0 17.1 Humphrey, Peter A; Dehner, Louis P; Pfeifer, John D (2008). The Washington Manual of Surgical Pathology (1st ed.). Lippincott Williams & Wilkins. pp. 650. ISBN 978-0781765275.
- ↑ PST. 22 February 2010.
- ↑ Humphrey, Peter A; Dehner, Louis P; Pfeifer, John D (2008). The Washington Manual of Surgical Pathology (1st ed.). Lippincott Williams & Wilkins. pp. 651. ISBN 978-0781765275.
- ↑ PST. 14 February 2011.
- ↑ PST. 22 February 2010.
- ↑ URL: http://atlasgeneticsoncology.org/Tumors/Ewing5010.html. Accessed on: 23 February 2010.
- ↑ Turc-Carel C, Aurias A, Mugneret F, et al. (June 1988). "Chromosomes in Ewing's sarcoma. I. An evaluation of 85 cases of remarkable consistency of t(11;22)(q24;q12)". Cancer Genet. Cytogenet. 32 (2): 229–38. PMID 3163261.
- ↑ Papalas JA, Balmer NN, Wallace C, Sangueeza OP (June 2009). "Ossifying dermatofibroma with osteoclast-like giant cells: report of a case and literature review". Am J Dermatopathol 31 (4): 379-83. doi:10.1097/DAD.0b013e3181966747. PMID 19461244.
- ↑ Humphrey, Peter A; Dehner, Louis P; Pfeifer, John D (2008). The Washington Manual of Surgical Pathology (1st ed.). Lippincott Williams & Wilkins. pp. 638. ISBN 978-0781765275.
- ↑ URL: http://bestpractice.bmj.com/best-practice/monograph/780/basics/classification.html. Accessed on: 7 April 2011.
- ↑ Kumar, Vinay; Abbas, Abul K.; Fausto, Nelson; Aster, Jon (2009). Robbins and Cotran pathologic basis of disease (8th ed.). Elsevier Saunders. pp. 1247. ISBN 978-1416031215.
- ↑ URL: http://emedicine.medscape.com/article/1253223-overview. Accessed on: 6 January 2011.
- ↑ URL: http://www.wheelessonline.com/ortho/pigmented_villonodular_synovitis.
- ↑ URL: http://southbaypath.org/CaseImages/sb5260/sb5260.htm. Accessed on: 7 December 2010.
- ↑ Meydan N, Barutca S, Guney E, et al. (June 2006). "Brown tumors mimicking bone metastases". J Natl Med Assoc 98 (6): 950–3. PMC 2569361. PMID 16775919. http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2569361/?page=1.
- ↑ TN06 Emerg.
- ↑ Humphrey, Peter A; Dehner, Louis P; Pfeifer, John D (2008). The Washington Manual of Surgical Pathology (1st ed.). Lippincott Williams & Wilkins. pp. 648. ISBN 978-0781765275.
- ↑ Klatt, Edward C. (2006). Robbins and Cotran Atlas of Pathology (1st ed.). Saunders. pp. 420. ISBN 978-1416002741.